Carmen Nussbaum
ZMBH
Im Neuenheimer Feld 282
69120 Heidelberg, Germany
Tel.: + 49-6221 54 6825
Fax.: +49-6221 54 5894
c.nussbaum@zmbh.uni-heidelberg.de
PROPAGATION OF PROTEIN MISFOLDING IN NEURODEGENERATIVE DISEASES
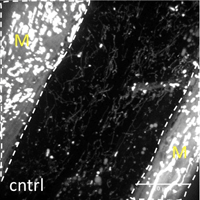
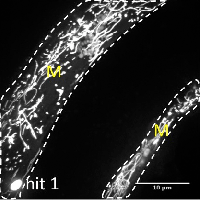
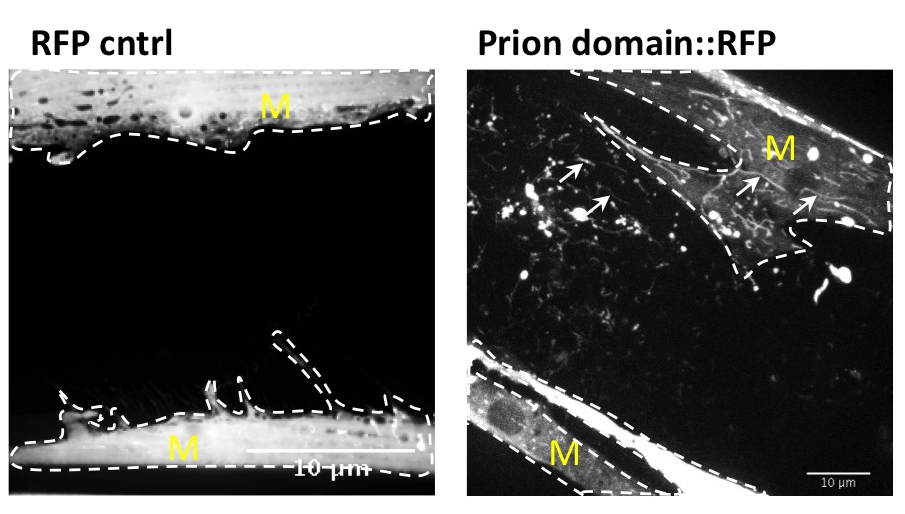
Many age-related neurodegenerative diseases, such as Alzheimer's disease or Parkinson's diseases, are characterized by deposits of aggregation-prone proteins. Proteinaceous "seeds" of specific disease-associated proteins can template the conversion of like proteins into pathogenic assemblies ranging from small oligomers to large amyloids. Moreover, protein misfolding seems to spread from an initial site of disease onset to interconnected brain regions during disease progression. This concept of induction and progressive spreading of protein misfolding has first been described for prions, the causative agent of transmissible spongiform encephalopathies (TSEs), but is now widely accepted as a unifying principle of many neurodegenerative disorders. Genetic screen for modifiers of prion-like spreading. C. elegans expressing fluorescently tagged prion-like proteins fed with different RNAi clones. In control animals, the protein is released from muscle cells (M) and taken up by neighboring cells. RNAi clones that interfere with intercellular spreading are classified as "hits". Muscle cell boundaries are outlined by dashed lines.
We are interested in how local protein misfolding is affecting neighboring cells and tissues and how proteostasis is orchestrated at the organismal level. How can misfolded proteins propagate within a cell? How are misfolded proteins transmitted between cells? Is there a correlation between aggregation, spreading and toxicity? And how is this linked to aging? To address these questions we are using genetics, live cell fluorescent imaging and biochemistry, mainly in our favorite model system, the nematode Caenorhabditis elegans. In addition, we will test if the genes and pathways identified in C. elegans are conserved, using mammalian tissue culture cells.
We have developed a prion model in C. elegans and showed that spreading of a prion domain occurs through the transport of autophagy-related lysosome-like vesicles between cells and tissues. This model offers great advantages over other animal models or tissue culture cells as intercellular transport of misfolded proteins is examined in an intact metazoan organism that is genetically tractable.
Selected Publications
Gao X, Carroni M, Nussbaum-Krammer C, Mogk A, Nillegoda NB, Szlachcic A, Guilbride DL, Saibil HR, Mayer MP, Bukau B. Human Hsp70 Disaggregase Reverses Parkinson's-Linked α±-Synuclein Amyloid Fibrils. Mol Cell. 2015 Sep 3;59(5):781-93.
Nussbaum-Krammer CI, Morimoto RI. Caenorhabditis elegans as a model system for studying non-cell autonomous mechanisms in protein-misfolding diseases. Dis Model Mech. 2014 Jan 7(1): 31-39.
Nussbaum-Krammer CI, Park K-W, Li L, Melki R, Morimoto RI. Spreading of a prion domain from cell-to-cell by vesicular transport in Caenorhabditis elegans. PLoS Genet. 2013 Mar;9(3):e1003351.
Krammer C, Kryndushkin D, Suhre MH, Kremmer E, Hofmann A, Pfeifer A, Scheibel T, Wickner R, Schätzl HM, Vorberg I. The yeast Sup35NM domain propagates as a prion in mammalian cells. Proc Natl Acad Sci U S A. 2009 Jan 13;106(2):462-7.
Krammer C, Schätzl HM, Vorberg I. Prion-like propagation of cytosolic protein aggregates: insights from cell culture models. Prion. 2009 Oct;3(4):206-12.