

Welcome to the Sasse lab!
|
|
Gene expression is the output of multiple processes that integrate transcriptional and post-transcriptional mechanisms. Transcription factors (TFs) bind specific sequence patterns in the DNA (aka motifs) to regulate transcriptional bursts of closeby or even distant genes. The fate of the generated mRNAs is determined by post-transcriptional factors, such as RNA binding proteins (RBPs, Sasse, Ray, Laverty et al. 2025), which bind to the produced transcript in a similar sequence-dependent manner to orchestrate a cascade of processes that lead to the translation of the mRNA into the final protein product. Cis-regulatory elements (CREs) in these genomic sequences determine gene expression levels across time and space. Modern genomic assays measure multiple regulatory events at cell type resolution. Genomic sequence-to-function (S2F) models can learn from collections of these rich data sources the relationship between sequence and the measured signal. These models are trained to take as input genomic DNA across the genome and predict as output experimental measurements such as chromatin accessibility (Chandra et al. 2025) or gene expression (Sasse, Ng, Spiro et al. 2023). S2F models are powerful tools for modern synthetic genomics because they allow us to: (1) query in-silico how arbitrary genetic variants impact gene expression (Tu, Sasse, Chowdhary et al. 2025), (2) gain biological insights into sequence determinants of gene expression (Sasse et al. 2024, iScience), and (3) in-silico design regulatory elements with specific cellular properties. The goal of our research is to develop accurate machine learning models that enable us to exploit large-scale gene expression and CRE datasets, to learn how CREs encode information about gene expression (Sasse, Chikina and Mostafavi, 2024). 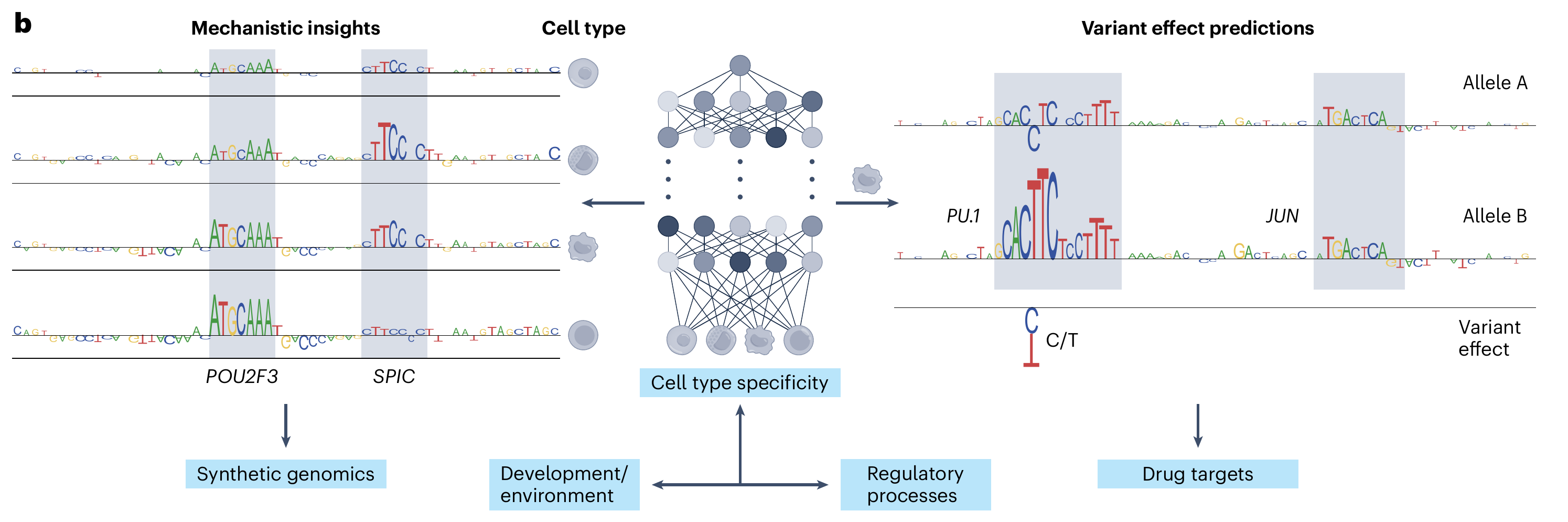
Sequence-to-function models take as input genomic DNA and learn to predict its functional properties such as gene expression in a cell-type-dependent and cell-state-dependent manner. Once trained, these models can be used to predict the impact of arbitrary genetic variations (right) and to derive biological insights into the sequence grammar that determines context-dependent gene regulation (left). Ac, acetylation; Me, methylation; Me3, trimethylation; RBPs, RNA-binding proteins; TAD, topologically associating domain. From (Sasse, Chikina, Mostafavi et al. 2024) We have open positions! Please see hereSelected publications
Tu, X.*, Sasse* A., Chowdhary K.*, et al. 2025. Deep Genomic Models of Allele-Specific Measurements. Preprint, bioRxiv, https://doi.org/10.1101/2025.04.09.648060. |